EMCO MAXXMILL 750
Begrüßen wir unser neues EMCO!
Die Konstruktion aus Gusseisen und geschweißtem Stahl garantiert ein Höchstmaß an Steifigkeit und Thermosymmetrie. Kurze Kraftflüsse sorgen für höchste Präzision und exzellente Oberflächenqualität des Werkstücks.
Mithilfe der 5-Seiten-Bearbeitung mit einem einzigen Aufstell und Schwenk-Rundtisch, können wir Teile in einem Klemmbereich von 750 x 600 mm fräsen.
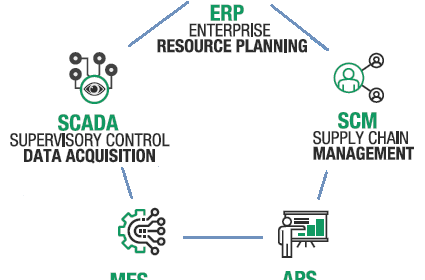
Dank der direkten Interaktion zwischen EMCO Apps und der Maschinensteuerung stehen alle Daten zur Produktivität auf einen Blick zur Verfügung: eine großartige Unterstützung in unserem Industrie 4.0-Ansatz!